


FDA Requirements for AI in Patient Engagement Tools

Key Takeaways
FDA Requirements for AI in Patient Engagement Tools
The FDA's approach to regulating AI in patient engagement tools focuses on risk management and ensuring safety. Tools are evaluated based on their intended use, clinical risk, and the significance of the information they provide. Here's what you need to know:
- Regulatory Pathways: AI tools are categorized as Class III (high-risk), De Novo (new moderate-risk), or 510(k) (low/moderate-risk with a similar existing device). Each pathway has specific approval requirements.
- Risk Assessment: The FDA uses the IMDRF framework to classify risks by healthcare context and the tool's role in clinical decision-making.
- Lifecycle Oversight: The FDA's Predetermined Change Control Plans (PCCPs) allow pre-approved AI updates, ensuring tools stay current without compromising safety.
- Transparency: Clear labeling and updated information on AI capabilities are mandatory to help clinicians and patients trust these tools. This is especially critical for an AI mentor designed to support patient journeys.
As of July 2025, the FDA had cleared 1,250 AI-enabled medical devices, emphasizing the growing importance of compliance with these regulations. Patient engagement tools must align with these standards to ensure they are safe, effective, and trusted by users.
1. FDA Regulatory Framework for AI Tools

Regulatory Pathways
The FDA organizes AI-powered patient engagement tools into three pathways, depending on their intended use and associated risk levels. High-risk tools, classified as Class III devices, require Premarket Approval (PMA) because they directly diagnose or treat medical conditions. Tools with low to moderate risk but without a legally marketed equivalent are evaluated under the De Novo Classification. Meanwhile, tools that are substantially similar to an already authorized device follow the Premarket Notification (510(k)) process.
For example, Apple used the 510(k) pathway to gain FDA clearance for its hypertension feature. These pathways guide the FDA's risk-based requirements for each category.
Risk-Based Requirements
The FDA uses the International Medical Device Regulators Forum (IMDRF) framework to determine risk levels for patient engagement tools. This framework evaluates two key factors: the healthcare situation (categorized as Critical, Serious, or Non-Serious) and the significance of the information provided (whether it Treats/Diagnoses, Drives Clinical Management, or Informs Clinical Management). Tools that are purely administrative often fall under the FDA's enforcement discretion.
"FDA's regulatory oversight applies to GenAI-enabled products that meet the definition of a device; such oversight is risk-based, taking into consideration the product's intended use and technological characteristics." – FDA
For instance, an app that summarizes interactions might be considered administrative, while one that provides a diagnosis would qualify as a regulated device.
Lifecycle Management
The FDA's Predetermined Change Control Plans (PCCP) simplify the process of updating AI models by allowing manufacturers to pre-authorize certain changes. This eliminates the need for a new marketing submission with every modification. The framework supports both continuous and manual updates, provided they align with the pre-approved plan.
"Obtaining FDA authorization of a PCCP as part of a marketing submission for an AI-DSF allows a manufacturer to modify its AI-DSF over time in accordance with the PCCP instead of obtaining separate FDA authorization for each significant change." – FDA
A valid PCCP must include three components: a Description of Modifications, a Modification Protocol (detailing how updates will be validated), and an Impact Assessment to evaluate the benefit–risk profile. Manufacturers are encouraged to use the Q-Submission Program to get FDA feedback on proposed PCCPs before submitting them formally.
Transparency Standards
Transparency is a cornerstone of the FDA's regulatory approach, designed to help clinicians make informed decisions. In June 2024, the FDA issued guiding principles for transparency in machine learning-enabled medical devices, emphasizing the importance of understanding a tool's limitations and the data used during its development. The FDA also maintains a public database of authorized AI-enabled devices.
For devices governed by PCCPs, the FDA requires that labeling be updated as the algorithm evolves, ensuring users are always aware of the tool's current capabilities and limitations. In April 2024, the FDA's digital health centers released a paper outlining plans to gather input on "explainability" and "governance" to further refine these transparency standards. This focus is particularly critical for Generative AI tools, where inaccuracies or "hallucinated" outputs could erode patient trust.
Together, these regulatory measures ensure that digital health tools balance safety, efficacy, and innovation in the rapidly evolving field of AI-driven patient engagement.
sbb-itb-8f61039
2. PatientPartner's Compliance-Ready Infrastructure
Regulatory Pathways
PatientPartner's platform is designed to align with FDA regulatory pathways, including 510(k), De Novo, and PMA, depending on the risk level and intended use of each tool. Its adaptable architecture ensures that the platform can meet the specific needs of pharmaceutical and med-tech partners, whether it's for tools aimed at monitoring treatment adherence or creating patient education programs.
The platform accommodates both Basic and Enhanced documentation levels. Enhanced documentation is required when software failure could lead to hazardous situations, such as a risk of death or serious injury. To meet these rigorous standards, PatientPartner integrates strong SDS and unit testing protocols into its framework.
This adaptable design lays the groundwork for effective lifecycle management.
Lifecycle Management
PatientPartner follows the FDA's guidelines for ongoing oversight by implementing a Total Product Lifecycle (TPLC) approach. This method covers every phase, from initial planning to real-world performance evaluation. By using this comprehensive process, the platform ensures that its AI-powered mentorship tools stay compliant and effective throughout their operational lifespan.
The platform also supports the FDA-approved PCCP, which allows for planned AI updates without requiring resubmission, as long as they adhere to an approved Modification Protocol. Built-in workflows within the platform ensure these updates meet PCCP standards.
Transparency Standards
PatientPartner prioritizes transparency in line with FDA principles, including the agency's June 2024 guidelines for machine learning-enabled medical devices. The platform documents critical data sources, model limitations, and performance metrics, providing clinicians with the information needed to make informed decisions. This transparency helps clarify how the mentorship system operates and the types of clinical decisions it can support.
Additionally, real-world performance monitoring is a key feature, tracking metrics like accuracy, efficiency, and user satisfaction in live environments. This ensures the platform consistently meets safety and effectiveness standards even after deployment.
FDA Guidance on Artificial Intelligence (AI) in Medical Devices
Pros and Cons
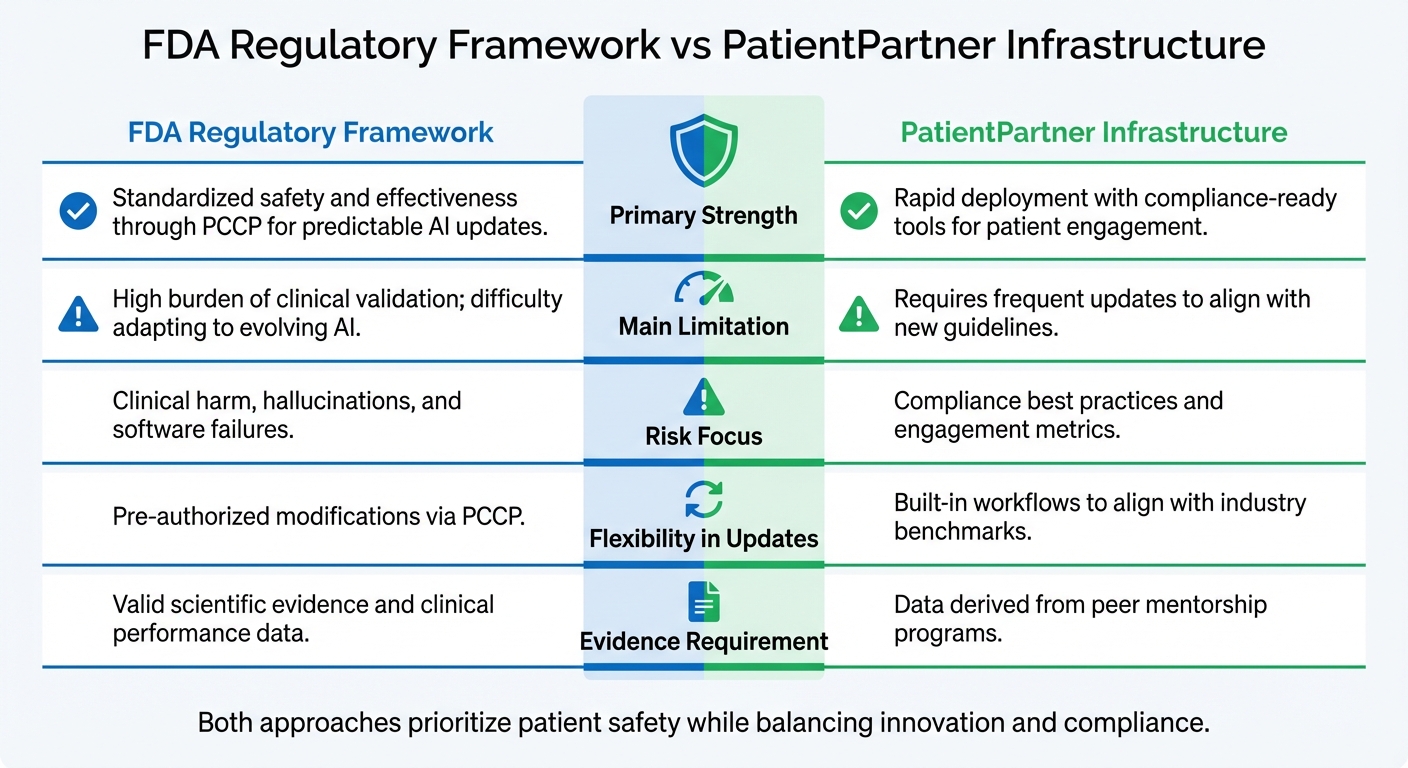
FDA vs PatientPartner: AI Patient Engagement Compliance Approaches
The FDA and PatientPartner each bring distinct strengths to the table when it comes to patient engagement, offering a balanced perspective for those considering AI implementation strategies.
The FDA's primary focus lies in ensuring clinical safety and maintaining standardized validation. For instance, on August 18, 2025, the agency finalized its guidance on Predetermined Change Control Plans (PCCP), which aims to support iterative innovation while upholding strict oversight. However, the FDA has openly acknowledged a key challenge:
"The FDA's traditional paradigm of medical device regulation was not designed for adaptive artificial intelligence and machine learning technologies".
This regulatory approach can create hurdles for AI tools that require frequent updates to stay relevant in a fast-paced technological landscape. On the other hand, PatientPartner strikes a balance between compliance and agility, addressing evolving market demands more seamlessly.
PatientPartner's platform is designed for speed and practical application. It provides compliance-ready templates that integrate industry benchmarks, enabling companies to enhance patient engagement without having to construct regulatory frameworks from scratch. The platform emphasizes measurable outcomes, such as improved treatment adherence and patient adoption, achieved through structured peer-to-peer mentorship programs. Additionally, PatientPartner stays proactive by updating its processes to align with new guidelines, like the January 7, 2025 draft on Artificial Intelligence-Enabled Device Software Functions: Lifecycle Management.
When it comes to risk management, the two approaches diverge significantly. The FDA prioritizes preventing clinical harm, especially in the context of generative AI, where:
"Hallucinations, particularly those that may appear to be authentic outputs to users, may present a significant challenge".
In contrast, PatientPartner mitigates these risks by grounding its decisions in real-world mentorship, reducing the uncertainties associated with black-box AI systems.
The table below highlights the key differences between the FDA's regulatory framework and PatientPartner's infrastructure:
| Aspect | FDA Regulatory Framework | PatientPartner Infrastructure |
|---|---|---|
| Primary Strength | Standardized safety and effectiveness through PCCP for predictable AI updates | Rapid deployment with compliance-ready tools for patient engagement |
| Main Limitation | High burden of clinical validation; difficulty adapting to evolving AI | Requires frequent updates to align with new guidelines |
| Risk Focus | Clinical harm, hallucinations, and software failures | Compliance best practices and engagement metrics |
| Flexibility in Updates | Pre-authorized modifications via PCCP | Built-in workflows to align with industry benchmarks |
| Evidence Requirement | Valid scientific evidence and clinical performance data | Data derived from peer mentorship programs |
Conclusion
Integrating AI-powered patient engagement tools with FDA regulations is all about creating systems that both patients and providers can trust. The FDA’s framework ensures "reasonable assurance of safety and effectiveness" by demanding thorough verification and validation before AI algorithms are deployed in healthcare settings. Without these safeguards, risks like hallucinations and data drift could jeopardize patient safety.
As of July 2025, the FDA had cleared over 1,250 AI-enabled medical devices, a jump from 950 in August 2024. This rapid expansion underscores the importance of tools like Predetermined Change Control Plans (PCCPs), which allow manufacturers to outline AI updates in advance while adhering to safety protocols.
To navigate this evolving landscape, there are a few key steps to consider. Start by engaging with the FDA early through the Q-Submission Program to get feedback on your PCCPs and clarify whether your tool falls under "significant risk" criteria. Next, develop robust monitoring systems to identify data drift and performance issues as soon as they arise, minimizing inaccuracies in clinical settings. Finally, focus on transparency - use plain-language explanations to show how AI impacts healthcare decisions. This openness fosters trust, an essential ingredient for long-term patient engagement. Balancing these regulatory demands with innovation is critical for success.
PatientPartner offers a great example of how to apply these principles effectively. By emphasizing real-world mentorship instead of relying on opaque AI models, the platform meets regulatory standards while staying nimble enough to engage patients meaningfully. This approach aligns seamlessly with the FDA's Total Product Life Cycle (TPLC) framework.
"AI technologies have the potential to transform healthcare by deriving new and important insights from the vast amount of data generated during the delivery of healthcare every day".
Realizing this potential means striking the right balance - pushing innovation forward while upholding the rigorous safety measures needed to protect patients and ensure fair outcomes for all.
FAQs
What FDA regulations apply to AI tools used in patient engagement?
The FDA oversees AI-powered tools for patient engagement by evaluating their intended use and associated risk. If a tool qualifies as a Software as a Medical Device (SaMD), it must go through one of the FDA’s established approval pathways: 510(k) clearance, De Novo classification, or Premarket Approval (PMA). These processes ensure the tool adheres to required safety and performance standards.
For AI tools deemed lower risk and not classified as medical devices, the FDA often applies enforcement discretion. This means these tools can function with fewer regulatory hurdles, striking a balance between encouraging innovation in patient engagement and safeguarding patient safety and confidence.
What is the FDA's Predetermined Change Control Plan (PCCP), and how does it support AI tool updates?
The FDA's Predetermined Change Control Plan (PCCP) offers a way for AI-powered tools to secure pre-approval for specific software updates before they’re even needed. This forward-thinking approach allows updates to roll out faster and more efficiently, all while ensuring safety and performance standards remain intact.
By simplifying the regulatory process, the PCCP enables developers to stay compliant while making ongoing improvements to their tools. This is especially important for AI applications in critical areas like patient engagement and adherence programs, where consistent functionality can make a real difference.
Why is transparency crucial for AI tools used in patient engagement?
Transparency plays a key role in making sure AI-driven tools for patient engagement are safe, effective, and reliable. When developers openly share details about how their AI models are built, tested, and monitored, it helps everyone - regulators, healthcare providers, and patients - gain a clearer understanding of how these tools operate. This allows for more informed decision-making.
Being transparent also aligns with FDA compliance standards, minimizes potential performance issues, and strengthens trust in the tool's ability to support long-term patient commitment and better health outcomes. In sensitive healthcare environments, trust is everything, and openness is the foundation of that trust.
Author

